欧盟体外诊断医疗器械第2017/746号法规
第2017/746号法规 (IVDR法规) 是体外诊断医疗器械监管的一个新的里程碑。通过其113篇条例和15个附录,体外诊断医疗器械法规为医疗器械制造商在监管方面带来了众多新的挑战。
体外诊断医疗器械法规 (IVDR) 要求所有体外诊断医疗设备制造商全面修订其质量管理体系和所有产品的技术文档。生产商们必须实施新流程来应对不断增加的监管要求。其中, 医疗器械唯一器械标识,性能评估和上市后监管等主题仅仅是医疗设备行业中即将到来的众多变化的冰山一角。
第2017/745号和2017/746号法规的比较
通过直接比较可看到这两版法规具有很大的相似性。在适用范围方面,MDR法规中对于医疗器材80%的规定都适用于IVDR法规对体外诊断器材的要求,此外,两者的章节结构和内容要求均可以1:1的方式转换。
与MDR相比,IVDR内容的最大差异在于分类系统 (第47,48条,附录VIII-XI),性能评估 (第56条,附录XIII) 以及对性能研究的要求 (第57,58条,附录XIII)。
就2017/746与2017/745法规间的差异我们为您做了一份详细的对比。您可以在我们的下载区下载该文档。
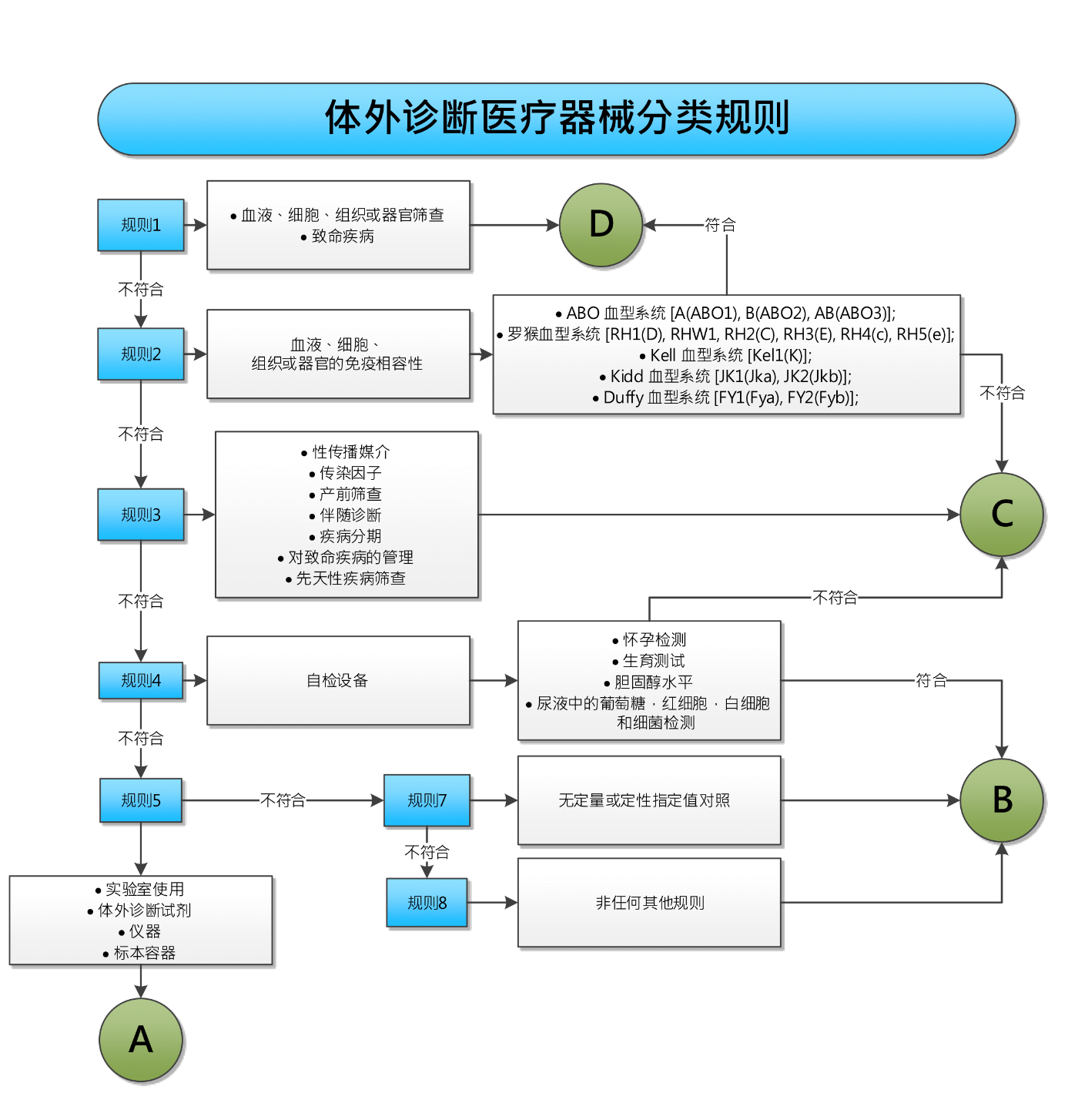
体外诊断医疗器械的风险分类系统
2017/746号法规从根本上改变了体外诊断医疗器械的风险分类系统。根据欧盟98/79/ EC指令附录II,体外诊断产品被分类为清单A所列器械 (风险最高),清单B所列器械 (高风险) 以及自用试验器械或其他体外诊断器械。对于未在附录II中提及的产品,并且非自用试验器械的其他体外诊断器械,认证机构无需对其进行符合性评估。
2017/746号法规将用基于规则的分类系统取代原基于清单的分类系统。体外诊断器械将基于七项特定规则被分为四个不同的风险等级 (A,B,C,D)。风险等级D用于描述最高风险等级。基于新的分类规则,预计约80%的产品将被划分为高于风险等级A的分类,因此这些产品必须由认证机构的进行符合性评估。2017/746号法规通过对制造商提高体外诊断器械的分类级别要求,不仅增加了文档记录的工作量,而且还增加了制造商对认证机构的依赖性。
性能评估
性能评估是指对数据进行评估和分析,以确定或验证体外诊断器械产品的科学有效性、分析性能和临床性能。
性能评估中所提供的临床证据将科学地证明所涉及的体外诊断器械产品可达到预期的临床用途,同时确认该产品的安全性。
有关体外诊断器械产品的科学有效性,分析性能和临床性能的数据,数据的评估以及由此得出的临床证据将被记录在性能评估报告中,性能评估报告也属于技术文档。
制造商在器械产品上市后应履行上市后性能跟踪调研计划和上市后监管计划。此过程中生成的数据将用于对体外诊断器械产品的性能评估以及相关文件在整个产品生命周期内的更新。
性能研究
性能研究是指为建立或确认体外诊断器械的分析性能或临床性能而进行的研究。
分析性能是指体外诊断器械正确检测或测量特定分析物的能力。临床性能是指体外诊断器械获得特定的临床状况相关的结果的能力。
体外诊断器械的性能研究将受到严格的监管,并且需要得到道德委员会以及高级联邦主管机构的批准。为申请性能研究,申请方需要准备一系列的相关文件;而申请的基础便是性能研究计划。该计划应说明临床性能研究的基本原理,目标,设计和所建议的分析,方法,监测和实施,以及它的记录要求。性能研究的结果必须展示在一份综合报告中。
您的产品是否已符合第2017/746号法规的要求? 您是否已根据第2017/746号法规的要求对您的产品进行了分类?你的性能评估是否同步于最新的法规要求?
我们不仅能在2017/746法规的落实过程中为您提供支持,并且能帮助您落实质量管理体系以及更新您的技术文档。我们可以为您展示2017/746法规关于性能评估提出的要求,此外,按照您的需要,我们还可以为您创建性能评估报告。
请您充分利用PROSYSTEM的专业经验和业内网络,使新版法规不会成为您和您公司的“不速之客”。
为确保您和您的员工能够在短时间内理解和掌握“医疗器械设备条例”中新增的和修订过的内容,您可以为贵公司预订我们的公开研讨会; 当然, 我们也可以为您量身定制培训课程。 通过研习实际案例和直接为您提供解决方案,我们将向您详细展示如何成功地计划至实现“体外诊断医疗器械法规“的要求。
如果您想了解更多有关2017/746法规的详细信息或我们的非约束性报价,请联系我们。