In Vitro Diagnostic – Regulation (EU) 2017/746
Regulation (EU) 2017/746 (IVDR) sets a new milestone in the regulation of in vitro diagnostic medical devices. In 113 articles and 15 annexes, the IVDR can create new regulatory challenges for IVD manufacturers.
The IVDR requires in-vitro diagnostics manufacturers to fully revise their quality management system and technical documentation for all products. New processes have to be implemented to address the increasing requirements. Topics such as Unique Device Identification, performance assessment or post-market surveillance are just a few examples of a variety of emerging changes in the medical device industry.
Comparing Regulation (EU) 2017/745 and 2017/746
These regulations are quite similar. Approximately 80% of the requirements apply to both medical devices as defined in Regulation 2017/745 and in-vitro diagnostics within the scope of Regulation 2017/746. Both the chapter structure and content requirements were transferred 1:1.
The greatest differences in content lie in the classification system (Articles 47-48, Annex VIII-XI), in the performance evaluation (Article 56, Annex XIII) and in the requirements for performance studies (Articles 57-58, Annex XIII).
We have detailed the differences between Regulation (EU) 2017/746 and Regulation (EU) 2017/745 The comparison is available in our download area.
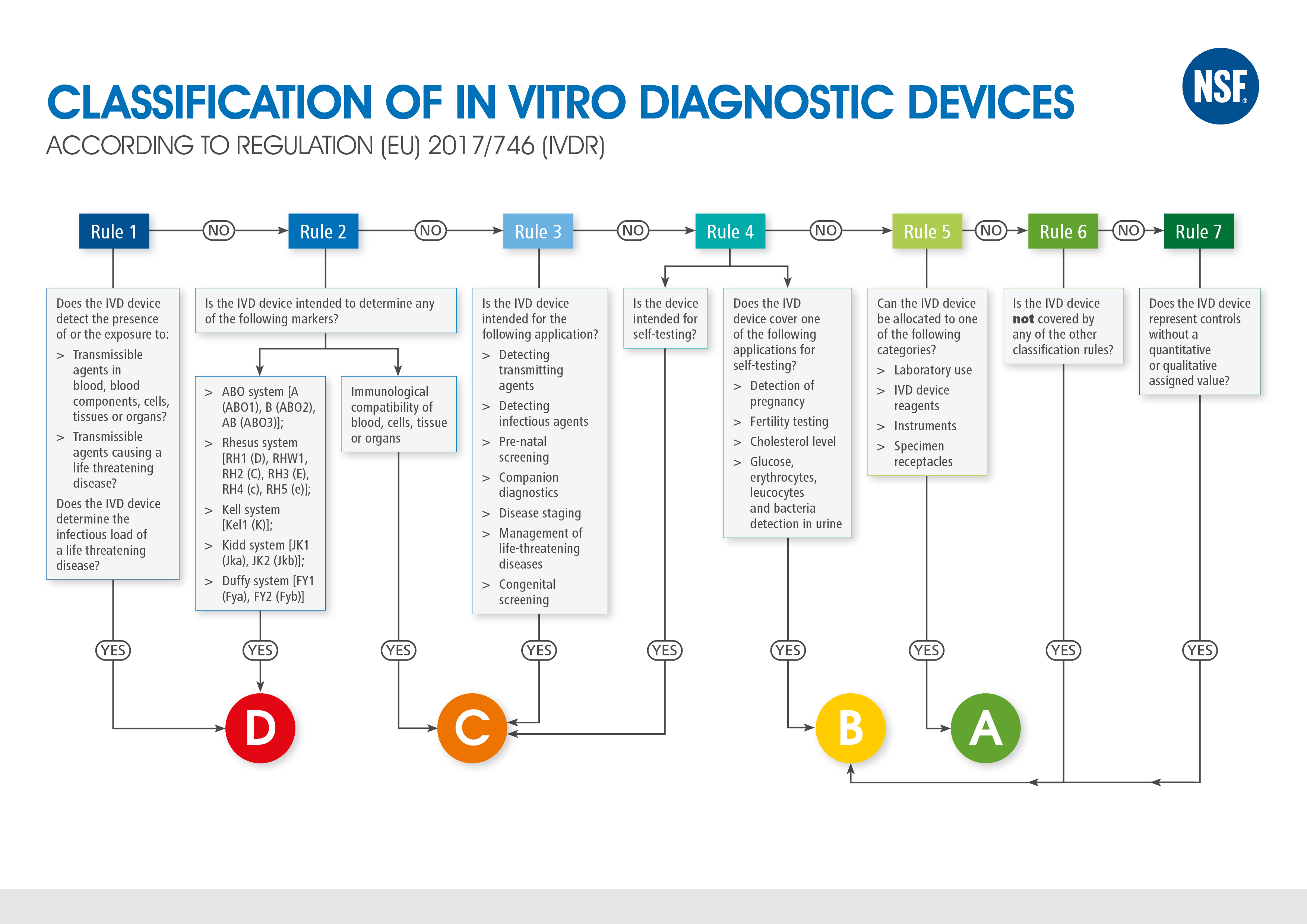
Classification of In Vitro Diagnostic Device
Regulation (EU) 2017/746 fundamentally changes the classification of in vitro diagnostic medical devices. In accordance with Directive 98/79/EC Annex II, in vitro diagnostic products were classified as products of list A (highest risk), list B (high risk), self-testing or other in vitro diagnostic products. For in vitro diagnostics not specified in Annex II or intended for self-application, no notified body had to be consulted for conformity assessment.
Regulation (EU) 2017/746 replaces the list-based classification system with a rule-based system. In the future, IVDs will be assigned to four different risk classes (A, B, C or D) based on seven specific rules. Risk class D describes the highest risk. Based on the new classification rules, it is expected that approximately 80% of all products will be assigned to a risk class higher than class A and will therefore be subject to a notified body’s conformity assessment. The new requirements of the regulation, which are also mandatory for manufacturers of products with the higher classification, demand not only a higher documentation effort, but also the involvement of a notified body.
Performance Evaluation
Performance evaluation means an assessment and analysis of data to establish or verify the scientific validity as well as the analytical and, where applicable, the clinical performance of a device
The clinical evidence to be provided by the performance evaluation will scientifically prove that the intended clinical benefit is achieved, and the device is safe.
The scientific validity data, analytical performance data and clinical performance data, their assessment and the clinical evidence derived therefrom, must be documented in the performance evaluation report, which must be part of the technical documentation.
The performance evaluation and supporting documentation must be updated throughout the lifecycle of the product based on the data resulting from the implementation of the post-market performance follow-up (PMPF) plan and the post-market surveillance plan.
Performance Study
A performance study is a study undertaken to establish or confirm the analytical or clinical performance of a device.
Analytical performance is the ability of a product to correctly detect or measure an analyte, and to yield results that are correlated with a clinical condition or a physiological or pathological process or state in accordance with the target population and intended user.
Performance studies are subject to strict regulatory requirements and require approval by an ethics committee as well as the competent federal supervisory authority. Several different documents must be prepared for the application based on the study plan. This plan sets out the rationale, objectives, design and proposed analysis, methodology, monitoring and performance of a clinical performance study, as well as the recordkeeping. The results of the study must be presented in a comprehensive report.
Do you comply with the requirements of Regulation (EU) 2017/746? Have you already classified your products in accordance with the requirements of Regulation (EU) 2017/746? Is your performance evaluation up to date?
We support you in implementing the requirements of Regulation (EU) 2017/746 both for implementation in your quality management system and for updating your technical documentation. We will show you the performance evaluation requirements and, if desired, also prepare your performance evaluation report.
Take advantage of our experience and network to ensure that the regulation does not become a surprise for you and your IVD products.
You and your employees must deal promptly with the new and changed requirements of the regulation. Book one of our seminars or a product-specific training. Based on practical examples and suggested solutions, we show you what successful planning for the implementation looks like. For further details on In Vitro Diagnostic – Regulation (EU) 2017/746 or for a no-obligation quote, please contact us.