In-vitro-Diagnostika – Verordnung (EU) 2017/746
Die Verordnung (EU) 2017/746 (IVDR) beschreibt einen neuen Meilenstein in der Regulierung von In-vitro-Diagnostika. In 113 Artikeln und 15 zusätzlichen Anhängen stellt die IVDR Medizinproduktehersteller vor neue regulatorische Herausforderungen.
Die IVDR fordert alle In-vitro-Diagnostika-Hersteller dazu auf, ihr Qualitätsmanagementsystem sowie die Technische Dokumentation sämtlicher Produkte vollständig zu überarbeiten. Neue Prozesse müssen implementiert werden, um die steigenden Anforderungen zu adressieren. Themen wie Unique Device Identification, Leistungsbewertung oder Post-Market Surveillance sind nur Beispiele einer Vielzahl aufkommender Veränderungen in der Medizintechnikindustrie.
Verordnung 2017/745 und 2017/746 im Vergleich
Beide Verordnungen zeigen im direkten Vergleich große Gemeinsamkeiten auf. Circa 80% der Anforderungen gelten sowohl für Medizinprodukte gemäß Geltungsbereich der Verordnung 2017/45 als auch für In-vitro Diagnostika im Geltungsbereich der Verordnung 2017/746. Sowohl die Kapitelstruktur als auch inhaltliche Anforderungen wurden 1:1 übertragen.
Die größten inhaltlichen Unterschiede liegen im Klassifizierungssystem (Art. 47, 48, Annex VIII-XI), in der Leistungsbewertung (Art. 56, Annex XIII) sowie in den Anforderungen an Leistungsstudien (Art. 57, 58, Annex XIII).
Alle Unterschiede der Verordnung 2017/746 im Vergleich zur Verordnung 2017/745 haben wir für Sie im Detail herausgestellt, die Sie in unserem Downloadbereich einsehen können.
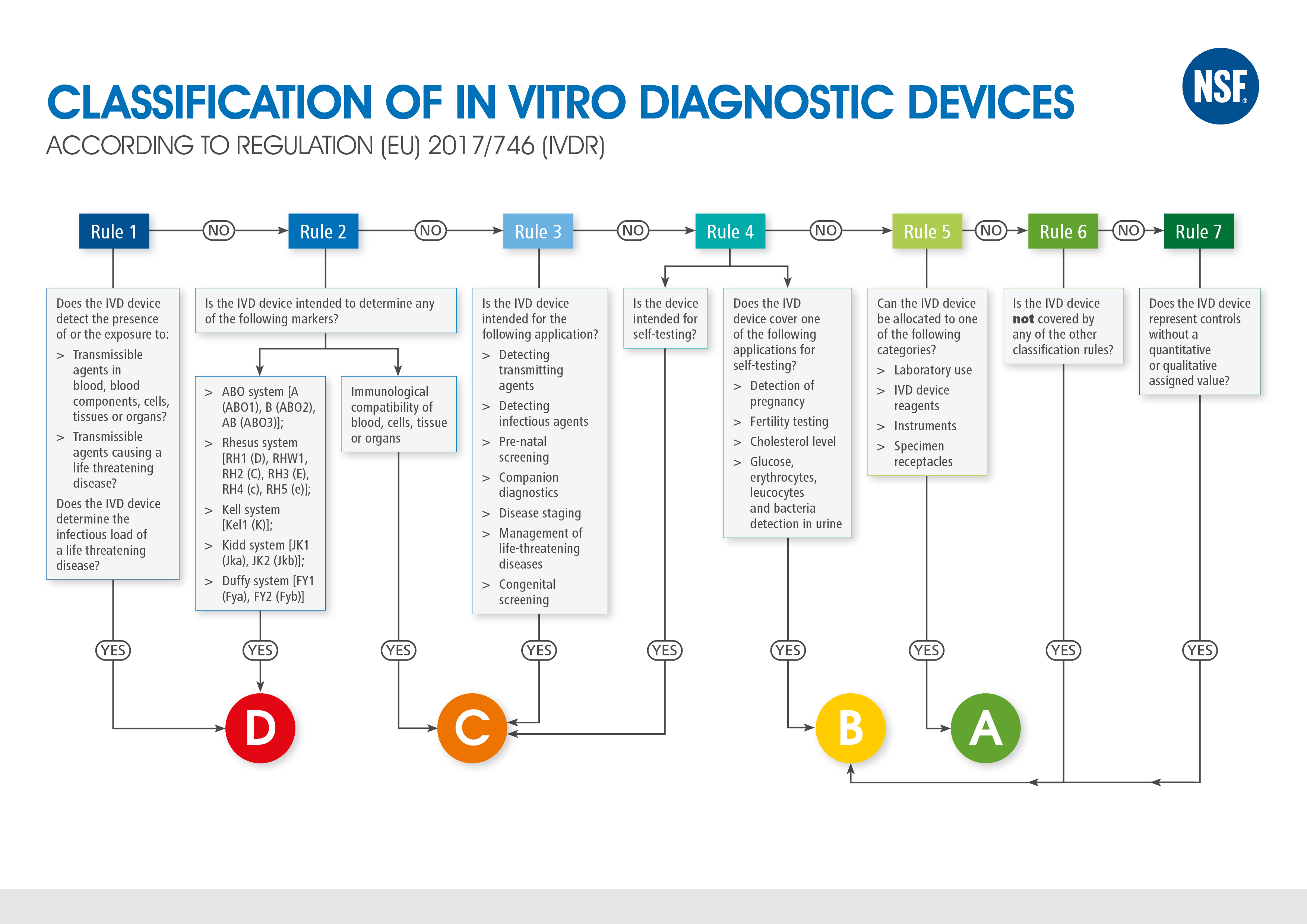
Klassifizierung von In-vitro Diagnostika
Die Verordnung (EU) 2017/746 ändert die Klassifizierung von In-vitro Diagnostika grundlegend. Gemäß Richtlinie 98/79/EG Anhang II wurden In-vitro Diagnostika unterschieden in Produkte der Liste A (höchstes Risiko), Produkte der Liste B (hohes Risiko), Produkte zur Eigenanwendung oder sonstige In-vitro-Diagnostika. Für sonstige In-vitro Diagnostika, die weder in Anhang II genannt noch zur Eigenanwendung bestimmt sind, musste keine Benannte Stelle zur Konformitätsbewertung einbezogen werden.
Im Rahmen der Verordnung (EU) 2017/746 wird das listenbasierte Klassifizierungssystem gegen ein regelbasiertes System ausgetauscht. Zukünftig werden IVDs anhand von sieben spezifischen Regeln vier verschiedenen Risikoklassen (A, B, C, D) zugeordnet. Die Risikoklasse D beschreibt das höchste Risiko. Aufgrund der neuen Klassifizierungsregeln wird erwartet, dass circa 80 % aller Produkte einer Risikoklasse höher als Klasse A zugeordnet werden und somit der Konformitätsbewertung einer Benannten Stelle unterstehen. Die neuen Anforderungen der Verordnung, die auch im Zuge der Höherklassifizierung an die Hersteller gestellt werden, fordern nicht nur einen höheren Dokumentationsaufwand, sondern auch die Abhängigkeit zu einer Benannten Stelle.
Leistungsbewertung
Die Leistungsbewertung bezeichnet eine Beurteilung und Analyse von Daten zur Feststellung oder Überprüfung der wissenschaftlichen Validität, der Analyseleistung und gegebenenfalls der klinischen Leistung eines Produkts.
Mit dem klinischen Nachweis, der durch die Leistungsbewertung erbracht werden soll, wird wissenschaftlich bewiesen, dass der beabsichtigte klinische Nutzen erreicht wird und das Produkt sicher ist.
Die Daten zur wissenschaftlichen Validität, zur Analyseleistung und zur klinischen Leistung, ihre Bewertung und der daraus abgeleitete klinische Nachweis werden in dem Bericht über die Leistungsbewertung dokumentiert. Der Bericht über die Leistungsbewertung ist Teil der technischen Dokumentation.
Die Leistungsbewertung und die dazugehörigen Unterlagen sind während des gesamten Lebenszyklus des Produkts anhand der Daten zu aktualisieren, die sich aus der Durchführung des Plans für die Nachbeobachtung der Leistung nach dem Inverkehrbringen des Herstellers (englisch „Post-Market-Performance-Follow Up“, PMPF) und dem Plan zur Überwachung nach dem Inverkehrbringen ergeben.
Leistungsstudien
Eine Leistungsstudie bezeichnet eine Studie zur Feststellung oder Bestätigung der Analyseleistung oder der klinischen Leistung eines Produkts.
Die Analyseleistung ist die Fähigkeit eines Produkts, einen bestimmten Analyten korrekt nachzuweisen oder zu messen. Die klinische Leistung ist die Fähigkeit eines Produkts, Ergebnisse zu liefern, die mit einem bestimmten klinischen Zustand korrelieren.
Leistungsstudien unterliegen strengen regulatorischen Anforderungen und bedürfen der Freigabe durch eine Ethikkommission sowie der zuständigen Bundesoberbehörde. Zur Antragstellung sind eine Reihe unterschiedlicher Dokumente vorzubereiten. Die Grundlage dafür stellt der Studienplan dar. In diesem Plan sind Begründung, Ziele, Konzeption und vorgeschlagene Analyse, Methodik, Überwachung und Durchführung einer klinischen Leistungsstudie sowie die Aufzeichnungen darüber dargelegt. Die Ergebnisse der Studie müssen in einem umfangreichen Bericht dargestellt werden.
Erfüllen Sie bereits die Anforderungen der Verordnung 2017/746? Haben Sie bereits Ihre Produkte gemäß den Anforderungen der Verordnung (EU) 2017/746 klassifiziert? Ist Ihre Leistungsbewertung noch auf dem aktuellen Stand?
Wir unterstützen Sie bei der Implementierung der Anforderungen der Verordnung 2017/746 sowohl zur Umsetzung in Ihrem Qualitätsmanagementsystem als auch bei der Aktualisierung Ihrer Technischen Dokumentation. Wir zeigen Ihnen auf, welche Anforderungen an die Leistungsbewertung gestellt werden und erstellen auch auf Wunsch Ihren Bericht zur Leistungsbewertung.
Nutzen Sie die Erfahrungen und das Netzwerk von NSF PROSYSTEM, damit die Verordnung nicht zur „Überraschung“ für Sie und Ihre IVD-Produkte wird.
Sie und Ihre Mitarbeiter müssen sich zeitnah mit den neuen und geänderten Anforderungen der Verordnung auseinandersetzen. Um Sie bestmöglich bei allen normativen und regulatorischen Aufgabenstellungen und Fragen zu unterstützen, bieten wir Ihnen jetzt die Möglichkeit, diese Dienstleistungen auch stundenweise zu buchen.
Warum jetzt auch stundenweise?
Unsere Experten haben langjährige Erfahrung und können Ihnen im Vorfeld die richtige Herangehensweise und eine detaillierte Aufwandsabschätzung kostenfrei zuschicken.Für weitere Details zum Thema “In-vitro-Diagnostika – Verordnung (EU) 2017/746” oder für ein unverbindliches Angebot
kontaktieren Sie uns gerne.
Ihre Weiterbildung zum Thema IVDR
Nutzen Sie unsere große Auswahl an verschiedenen IVD Seminaren, um sich bestmöglich auf die kommenden Anforderungen vorzubereiten:
• Inhouse-Schulungen: Wir bieten spezifisch auf Ihre Produkte abgestimmte Schulungen an. Diese können sowohl bei Ihnen vor Ort als auch online stattfinden. Kontaktieren Sie uns für ein Angebot gerne unter TrainingcenterGermany@nsf.org
• Offene Seminare: Buchen Sie einen unserer Plätze für ein Online-Seminar oder Präsenzseminar, profitieren Sie vom Mengenrabatt und diskutieren Sie mit dem Referenten und anderen Herstellern, wie sie die neuen und geänderten Anforderungen effektiv im Unternehmen umsetzen können.
Wir freuen uns auf Ihre Teilnahme.