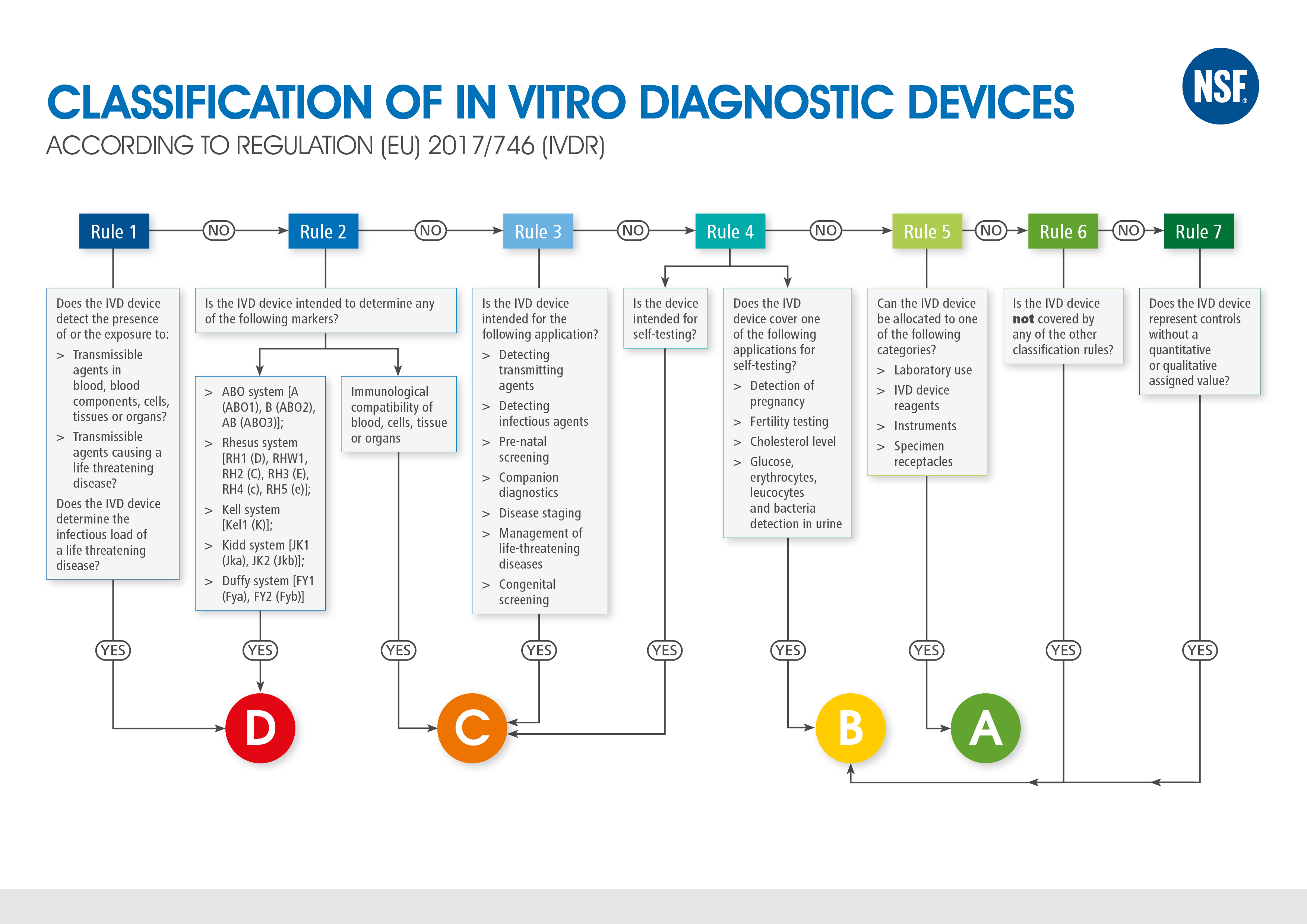
Die Verordnung (EU) 2017/746 ändert die Klassifizierung von In-vitro Diagnostika grundlegend. Gemäß Richtlinie 98/79/EG Anhang II wurden In-vitro Diagnostika unterschieden in Produkte der Liste A (höchstes Risiko), Produkte der Liste B (hohes Risiko), Produkte zur Eigenanwendung oder sonstige In-vitro-Diagnostika. Für sonstige In-vitro Diagnostika, die weder in Anhang II genannt noch zur Eigenanwendung bestimmt sind, muss keine Benannte Stelle zur Konformitätsbewertung einbezogen werden.
Im Rahmen der Verordnung (EU) 2017/746 wird das listenbasierte Klassifizierungssystem gegen ein regelbasiertes System ausgetauscht. Zukünftig werden IVDs anhand von sieben spezifischen Regeln vier verschiedenen Risikoklassen (A, B, C, D) zugeordnet. Die Risikoklasse D beschreibt das höchste Risiko. Aufgrund der neuen Klassifizierungsregeln wird erwartet, dass circa 80 % aller Produkte einer Risikoklasse höher als Klasse A zugeordnet werden und somit der Konformitätsbewertung einer Benannten Stelle unterstehen. Die neuen Anforderungen der Verordnung, die auch im Zuge der Höherklassifizierung an die Hersteller gestellt werden, fordern nicht nur einen höheren Dokumentationsaufwand, sondern auch die Abhängigkeit zu einer Benannten Stelle.
Haben Sie bereits Ihre Produkte gemäß den Anforderungen der Verordnung (EU) 2017/746 klassifiziert? Wir unterstützen Sie bei Ihrer Klassifizierung und der Kommunikation mit Ihrer Benannten Stelle und zeigen Ihnen auf, welche Anforderungen aufgrund der neuen Verordnung auf Sie zu kommen.